In recent years, the field of multiomics has undergone an increasing expansion of interest from the scientific community. Several techniques have been developed to gain an increase in resolution when studying cellular characteristics. Using multiomics technology, scientists can now rebuild cell lineage trajectories to further understand animal development, to unravel how cancer cells develop during carcinogenesis by linking mutations with cellular behavior, and to identify cell subtypes within a population of cells (Reviewed in 1).
Single-cell RNA-sequencing (scRNA-seq) saw the light of day in the 1990s when the groups of James Eberwine (University of Pennsylvania Medical School) and Gerard Brady (Ontario Cancer Institute) with respective colleagues used PCR and in vitro transcription at single cell level (2, 3).
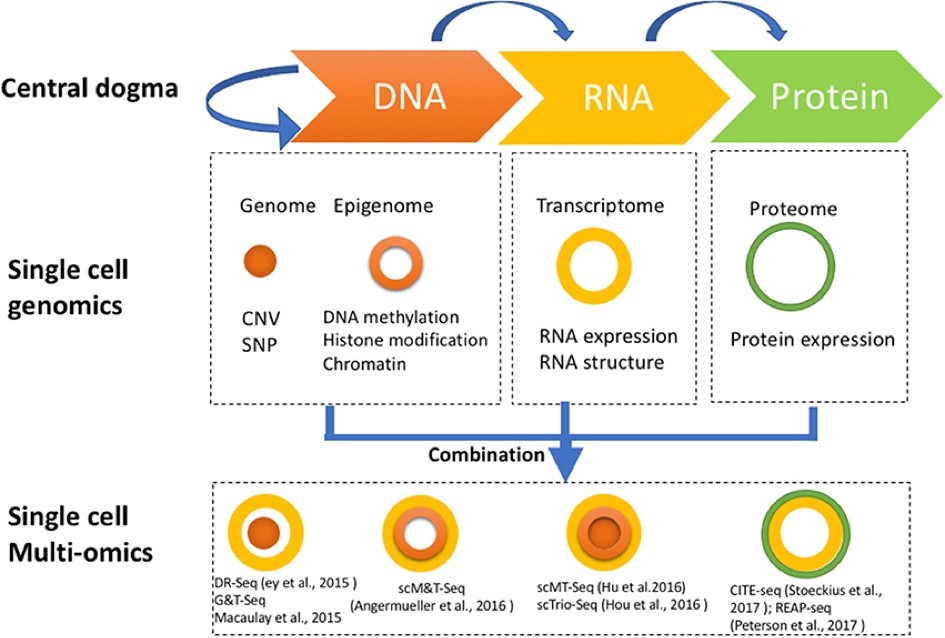
In more recent years, scRNA-seq was adapted and introduced to the omics era of the 21st century when Tang and colleagues exploited the possibilities of Next Generation Sequencing (NGS) for single cell characterization. This allowed them to discover several thousand more genes and unknown splice junctions using a single mouse blastomere than would have been possible with microarray technology, which would need hundreds of blastomeres (4). Since then, scRNA-seq technology has expanded rapidly with several different approaches that combine the pillars of the central dogma of molecular biology, as illustrated in Figure 1 (Reviewed in 1).
Figure 1. Diagram showing single cell genomics and single cell multi-omics in relation to the central dogma. Methods dealing with cellular genome and epigenome are represented by a filled orange sphere and an orange un-filled circle, respectively. Methods approaching the transcriptome are represented by a yellow circle and methods with a solution towards the proteome are represented by a green circle. CITE-seq and REAP-seq are the only two technologies aimed towards both the transcriptome and proteome. CNV, Copy Number Variation; SNP, Single Nucleotide Polymorphism. Image from Hu et al., 2018 (1).
In 2017, Stoeckius and colleagues published a paper describing a new method that could link data about the cellular transcriptome with expression levels of cell surface proteins. They named this method Cellular Indexing of Transcriptomes and Epitopes by Sequencing (CITE-seq) (5). At about the same time, Peterson and co-workers published a similar technique, which they named RNA Expression and Protein Sequencing Assay (REAP-seq) (6).
The CITE-seq and REAP-seq workflows offered a unique and much-awaited contribution to the multiomics field in that they are the first methods that link the expression of cell surface proteins with RNA expression levels.
CITE-seq
The central tool in the CITE-seq technique is an antibody that is conjugated to an oligonucleotide via a disulfide bond. This bond will break under reducing conditions, thus separating the antibody from the oligonucleotide. The oligonucleotide is composed of a PCR handle to initiate amplification. Downstream of the handle is a 15 bp barcode specific for identification of the antibody and a 3´ poly A tail for RNA capture (Figure 2) (5).
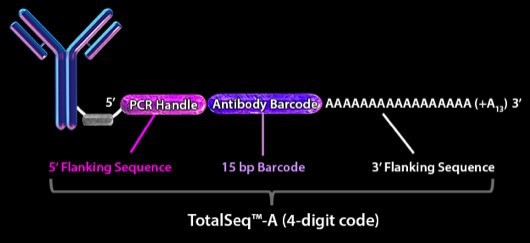
Figure 2. Schematic view of antibody bridged to oligonucleotide. The antibody is directed against a surface protein and is attached to an oligonucleotide consisting of a PCR handle in the 5´end that initiates a PCR reaction, an antibody barcode to distinguish a given antibody from other antibodies, and a poly-A tail for ligation to poly-dT primers on beads. Image from BioLegend, 2018.
Principle of the CITE-seq Workflow:
- The antibody complex is incubated together in a single cell suspension to allow antibodies to bind to their specific epitopes. This is followed by washing away of unbound excess antibodies (Figs. 3.1a and 3.1b, respectively).
- CITE-seq is designed to work with either the Drop-seq methodology (7) or using the Chromium system Single Cell 3´ Solution from 10x Genomics (10x Genomics, USA). Using the Drop-seq technique as an example, cells bound to antibodies and beads coated with poly-dT primers are separately pushed through a microfluidics device. Downstream, oil is entered into the microfluidics system thus creating nanoliter-sized droplets containing one bead and a cell-antibody-oligonucleotide complex (Fig. 3.2a).
- The cell is lysed in the droplet microenvironment, thus releasing the cellular mRNA contents, which then hybridize to the poly-dT primers on the bead surface (Fig. 3.2b).
- At this stage, the antibody is released from the oligonucleotide containing an antibody identification barcode and annealed to poly-dT primers on the bead (Fig. 3.2c). A reversed transcription amplification step is performed creating cDNA of intracellular mRNA origin (fig. 3.3a) and Antibody-Derived Tags (ADT) (Fig. 3.3b).
- The cDNA and ADTs are then size separated and converted into two Illumina-compatible sets of libraries, which are finally sequenced (Figs. 3.4 and 3.5) (5).
Multiplexing Possibilities Using Cell Hashing
Stoeckius and co-workers also developed a multiplexing strategy to distinguish between several samples in one pool by performing CITE-seq (5). This multiplexing strategy is referred to as cell “hashing” and utilizes a hashtag-oligo (HTO). The HTOs of the cell hashing technique differ in that they contain a modified PCR handle in comparison to the standard CITE-seq ADTs. This, in combination with the antibody directed against highly expressed omnipresent cell markers, allows multiplexing and subsequent differentiation of multiple samples in one CITE-seq experiment (8).
For human cells, the following immune markers are used: CD298, β2 microglobulin. For cells of mouse origin, immune markers CD45, and H-2 MHC class I are used (9). In addition to the two libraries generated by CITE-seq (cDNA of intracellular origin and ADTs, Figs. 3.3a and 3.3b), it is possible to generate a third library (HTO) when using cell hashing (8).
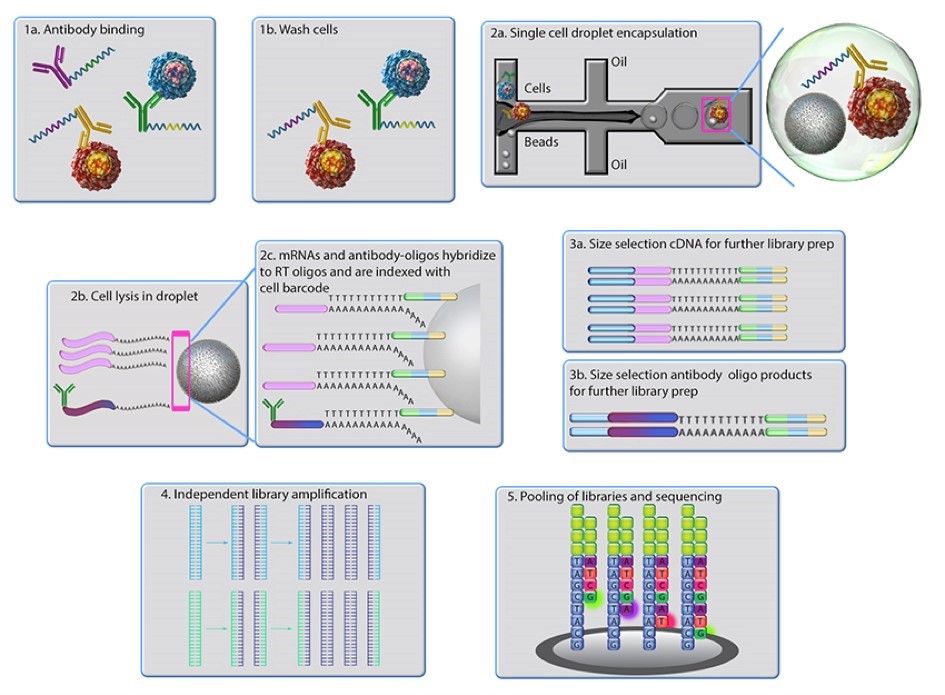
Figure 3. Principle of the CITE-seq technique. Antibodies conjugated with oligonucleotides are incubated with a sample of cells. The cell-antibody-oligonucleotide complex together with poly dT-coated beads is pushed into a microfluidics system. When oil is added downstream this creates a small vesicle containing one bead and one cell with an antibody-oligonucleotide complex bound to its surface. The cell is lysed and a reversed transcription of the mRNA to cDNA is performed thus creating cDNA of intracellular mRNA origin and Antibody Derived Tags (ADT). cDNA and ADTs are then converted into separate Illumina-compatible libraries. Image from BioLegend, 2018.
TotalSeq™-A from BioLegend
Recently, BioLegend together with New York Genome Center launched the CITE-seq and cell “hashing” techniques under the brand name TotalSeq™-A, designed specifically to work on Illumina sequencers and the 10x Genomics platform (9).
With this technology, we now have a very powerful proteogenomics tool at hand, and we are looking forward to see how it will advance basic and applied research in the future.
Could TotalSeq™ Be for You?
If you think TotalSeq™ might be the next big thing in your workflow, feel free to contact us at info@nordicbiosite.com for an informal chat and to learn more about what’s on offer. You can also read more about TotalSeq™-A on BioLegend’s website here.
References
Hu, Y. et al. Single Cell Multi-Omics Technology: Methodology and Application. Front. Cell Dev. Biol. 6: 28, 10.3389/fcell.2018.00028 (2018).
Brady, G., Barbara, M. & Iscove, N. N. Representative in vitro cDNA amplification from individual hemopoietic cells and colonies. Methods Mol. Cell Biol.2, 17-25 (1990).
Eberwine, J. et al. Analysis of gene expression in single live neurons. Proc. Natl. Acad. Sci. USA 89, 3010-3014 (1992).
Tang, F et al. mRNA-Seq whole-transcriptome analysis of a single cell. Nat Methods. May;6(5):377-82. doi: 10.1038/nmeth.1315 (2009).
Stoeckius, M. et al. Simultaneous epitope and transcriptome measurement in single cells. Nat Methods. Sep;14(9):865-868 (2017).
Peterson, V. et al. Multiplexed quantification of proteins and transcripts in single cells. Nat Biotechnol. Oct;35(10):936-939 (2017).
Macosko, E.Z. et al. Highly Parallel Genome-wide Expression Profiling of Individual Cells Using Nanoliter Droplets. Cell 161, 1202-1214 (2015).
[BioRxiv ahead of print] Stoeckius, M. et al. Cell “hashing” with barcoded antibodies enables multiplexing and doublet detection for single cell genomics. BioRxiv preprint first posted online Dec. 21, 2017; https://dx.doi.org/10.1101/237693doi (2017).
BioLegend (2018). Antibody Oligonucleotide Conjugation Services: TotalseqTM and CITE-seq Focus. Available at: https://www.BioLegend.com/totalseq [Accessed September 13, 2018].